Inferring evolutionary processes in populations of diverse ancestries
How do population histories influence genotypic and phenotypic variation? Answering this question relies on our ability to interpret the genetic signatures left by the timing and structure of demographic events. However, many population genetic models make a variety of simplifying assumptions. For example, while many classical models assume random mating, mating pairs are often found to be more similar in phenotype or genotype than expected under random mating.
|
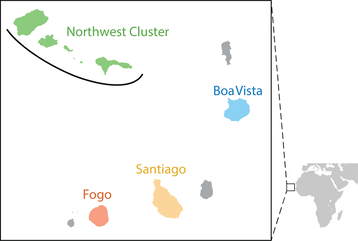
In my postdoctoral work in the Goldberg lab, we asked how mating patterns shape genetic variation and bias demographic inferences in the recently admixed human population of Cabo Verde (right). The islands of Cabo Verde were uninhabited prior to founding in the late 1400s. Cabo Verdeans today are admixed descendants of the Portuguese colonizers and enslaved West African people who founded the islands ~20-25 generations ago. By jointly considering the well-studied historical record of Cabo Verde and genome-wide SNP data, we find that sex-biased admixture and nonrandom mating shape genetic variation and influence demographic inference in this population, providing generalizable considerations for populations lacking historical records.
|
|
Recombination and chromosomal arrangement
|
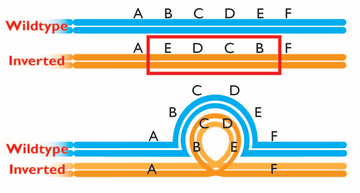
I am fascinated by chromosomal inversions and their roles in evolutionary processes. Inversions suppress the ability of recombination to shuffle combinations of alleles across loci, but we don’t fully understand how and to what extent. Though single-crossover products are prevented within inversion heterozygotes, non-crossover gene conversion and double crossovers can still occur. During my PhD, I aimed to improve our understanding of how recombination, particularly gene conversion, acts alongside natural selection to influence genome evolution and speciation.
|
|
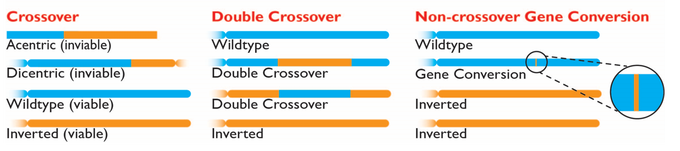
To better understand the recombination-barrier imposed by inversions, I examined rates of gene conversion in experimental crosses both within Drosophila pseudoobscura and between D. pseudoobscura and its naturally-hybridizing sister species D. persimilis. Through genome-wide empirical analyses of within species and in species hybrids, I observed that gene conversion rates in interspecies hybrids are at least as high as within-species estimates of gene conversion rates, and gene conversion occurs regularly within and around inverted regions of species hybrids, even near inversion breakpoints. This direct empirical assessment of gene conversion rates within inversions shows that gene conversion has the potential to reduce the efficacy of inversions as barriers to recombination over evolutionary time.
Improving methods in population genetics
|
Reproducibility in common population genetic summary statistics:
Dr. Kieran Samuk and I worked collaboratively to create software that overcomes common pitfalls in calculating nucleotide diversity within (π) and between (dxy) populations. Tools for calculating these common statistics often fail to distinguish missing data (sites and genotypes) from invariant data, leading to chronic underestimation of nucleotide diversity and a lack of reproducibility. To overcome these issues with a reproducible, transparent tool, we provide pixy. This software calculates π and dxy using VCFs containing invariant sites, missing sites, and missing genotypes. Accessibility of data and tools:
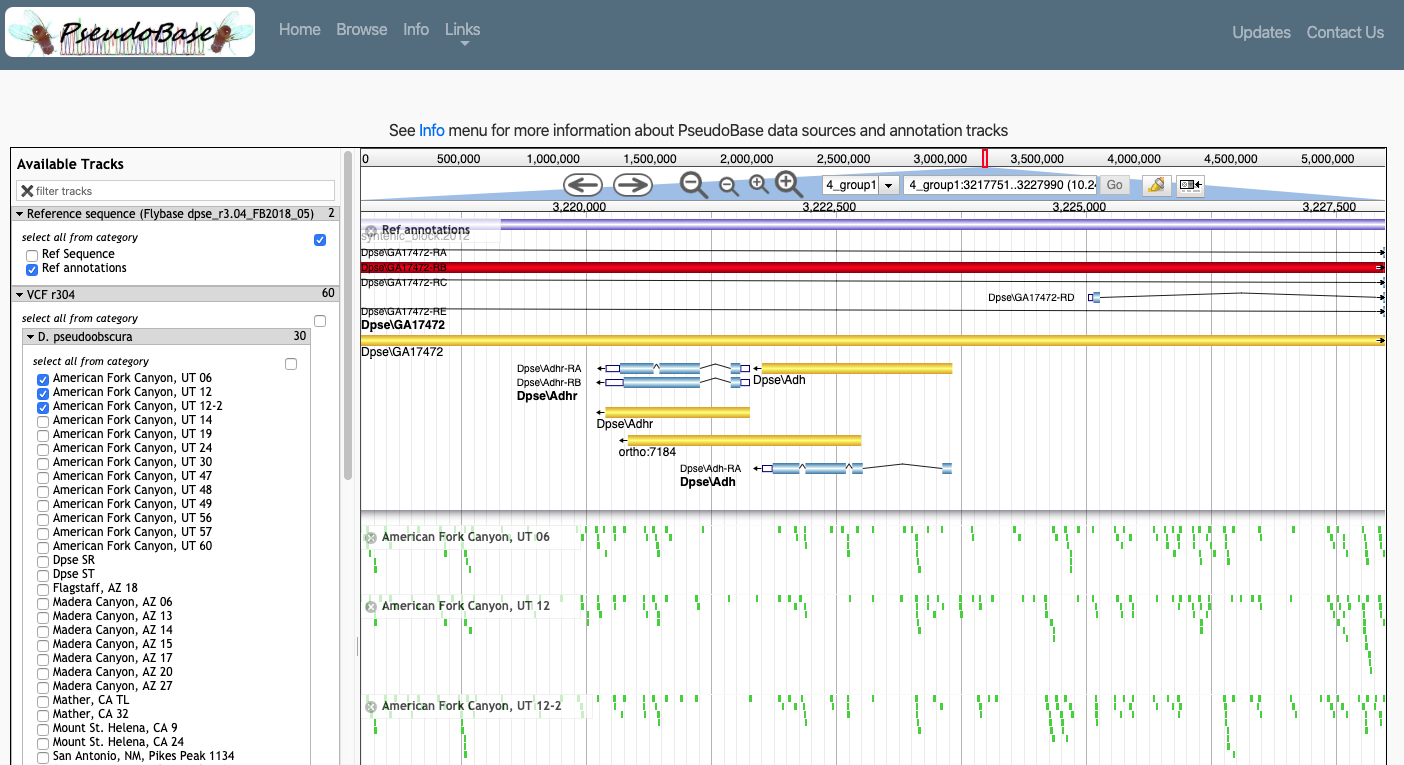
During my graduate studies, I worked with the Noor Lab to provide a community tool, PseudoBase 2.0, that lowers barriers to working with genomic data and facilitates the exploration of extensive data from the Drosophila pseudoobscura subgroup. PseudoBase couples these data with simple visualization and query tools via an intuitive graphical interface. We intend this to be a widely accessible tool particularly useful for pilot analyses, data checks, and educational purposes. More info can be found in our paper introducing PseudoBase. Leveraging ancestry to study evolutionary processes:
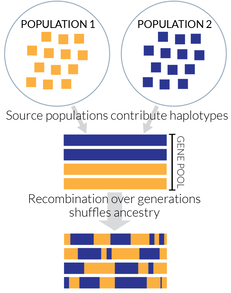
Recently admixed populations provide unique opportunities to study evolution on short timescales. When admixture introduces alleles at intermediate frequencies, this can bypass potential loss from drift and sometimes enable much faster adaptation compared to selection on new mutations, which is mutation‐limited. Thus, admixture is one of the fastest evolutionary processes to dramatically change the composition of a population, and ancestry patterns in admixed populations reveal changes on a much shorter timescale than allele frequency changes in the source populations. In the Goldberg Lab, we're working to create new statistical methods that leverage ancestry patterns in detecting and characterizing selection. Using ancestry, we aim to better understand adaptation, including cases where admixture can facilitate rapid adaptation when alleles from another population provide a selective advantage. |
|
The role of introgression in speciation and divergence
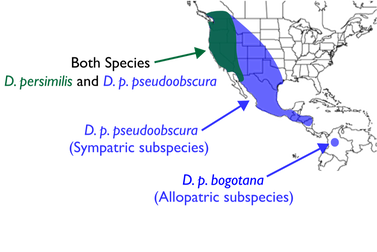
The sister species pair Drosophila pseudoobscura and D. persimilis present an opportunity to dissect an evolutionary history of divergence nuanced by multiple inversions, lineage sorting, and gene flow.
|
In collaboration with Carlos Machado and Mohamed Noor, I used whole-genome sequence data to explore patterns of introgression and divergence in the Drosophila pseudoobscura / D. persimilis system, with strategic use of an allopatric point of comparison, D. pseudoobscura bogotana (D. p. bogotana), which split from North American D. pseudoobscura (D. p. pseudoobscura) about 150,000 years ago. We examined the effects of introgression, but also evaluated how differences in evolutionary rate among lineages can influence patterns of divergence and tests for introgression. Consideration of ancestral inversion polymorphisms and different evolutionary rates in these populations revealed that recent gene exchange is a major but not the sole driver of variation in overall sequence divergence. Overall, we show that inversions have contributed to divergence patterns between Drosophila pseudoobscura and D. persimilis over three distinct timescales: 1) in the ancestral population, 2) during a period of ancient gene flow, and 3) during recent gene flow. These results represent progress towards a more cohesive understanding of how inversions shape gene flow at different timescales along the speciation continuum.
|
|
Toxicogenomics |
|
|
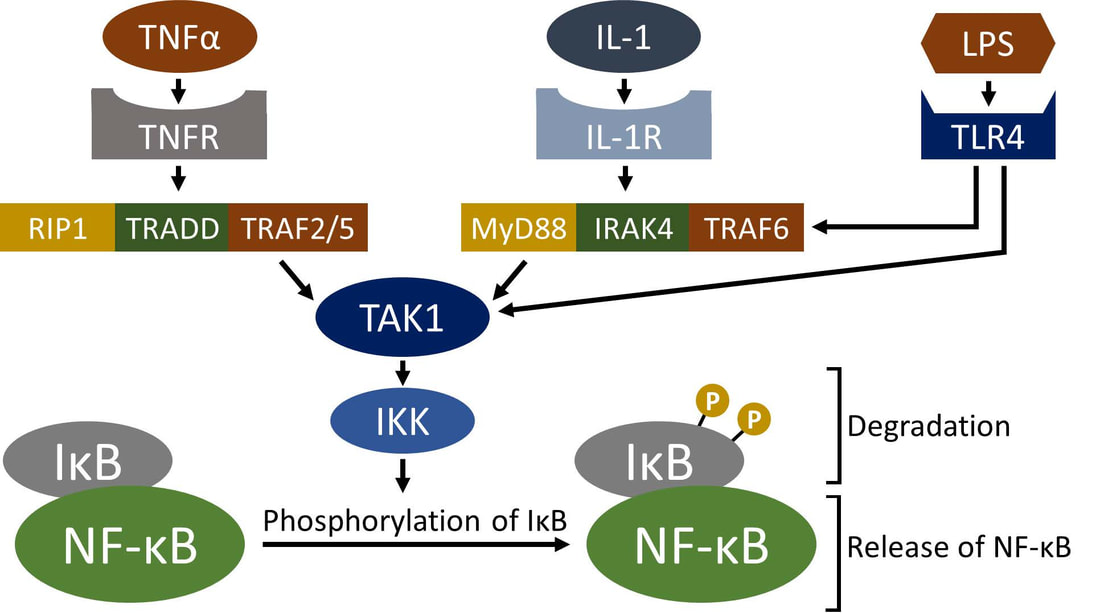
As a NSF graduate fellow, I collaborated with the lab of Dr. Chris Corton at the U.S. Environmental Protection Agency (EPA) to create and apply computational genomic strategies to predict mammalian responses to environmental chemicals. To protect people and ecosystems by regulating the tens of thousands of chemicals that are currently in commerce, the EPA is investigating gene expression profiling techniques that can readily interpret high throughput gene expression data. A critical gap in the application of gene expression profiling techniques is the availability of validated gene expression signatures (biomarkers) that have been robustly tested across laboratories, human cell culture models, gene expression platforms, and experimental designs. To address this gap, I built and validated a biomarker that predicts the perturbation of NFκB, a family of transcription factors with important roles in inflammation, immune response, and oncogenesis. This biomarker advances our understanding of this important transcription factor and provides a valuable tool for predicting NFκB modulation (associated manuscript currently in review). I also contributed R scripts to analyze data for other projects in the lab, such as for our recent publication in Toxicologic Pathology. This work was completed over a 4-month NSF-funded internship (Graduate Research Internship Program - GRIP).
|
|
